wisdom
Human variation
The difficulty of discovering links between genetic variants and phenotypic traits lies in the fact that there is a big intra-population genetic variability: a small subset of it will be related to the studied phenotype; the rest will just add noise. In consequence, understanding this variability is the first step in a genetic study. That is why, shortly after the human genome was sequenced, international projects aiming at characterizing divergences from the reference genome sprouted. Possibly the best-known are HapMap and 1000 Genomes. These projects use sequencing technologies at deep coverages to identify common and rare variants in the general population. They play two key roles in genetic studies: first, identifying the genome regions to survey; second, describing the population-specific variants.
The modern unit of genetic variation are single-nucleotide polymorphisms (SNPs) (Bush et al., 2012). SNPs are single base-pair changes in the DNA sequence that occur with high frequency in the human genome. They are, by far, the most common form of genetic variation in humans. SNPs usually involve two alleles, meaning that in a population there are two possible base-pairs for a genetic position. SNPs are characterized by their minor-allele frequency (MAF), that is, the frequency of the least common allele in the population.
Linkage disequilibrium
Linkage disequilibrium refers to the fact that contiguous SNP.
** how human variation changes from population to population **
Genome-wide association studies
The study of complex diseases, where multiple genetic factors contribute to the susceptibility, relies on the common disease, common variant} (CD/CV) hypothesis: common diseases are partly attributable to allelic variants present in more than 1-5% of the population, which cause, by themselves or in combinations, small increments in risk (1.1 - 1.5-fold). Due to this limited effect size, only partial correlations between causal variants and phenotypes are expected. This notion is radically different from rare diseases associated to rare variants with big effect sizes, where every carrier of the risk allele develops the disease (complete correlation). In fact, this is consistent with population genetic theory: variants that explain a large part of the disease by themselves would reduce the reproductive fitness of the individual and, in consequence, would be infrequent (Manolio et al., 2009).
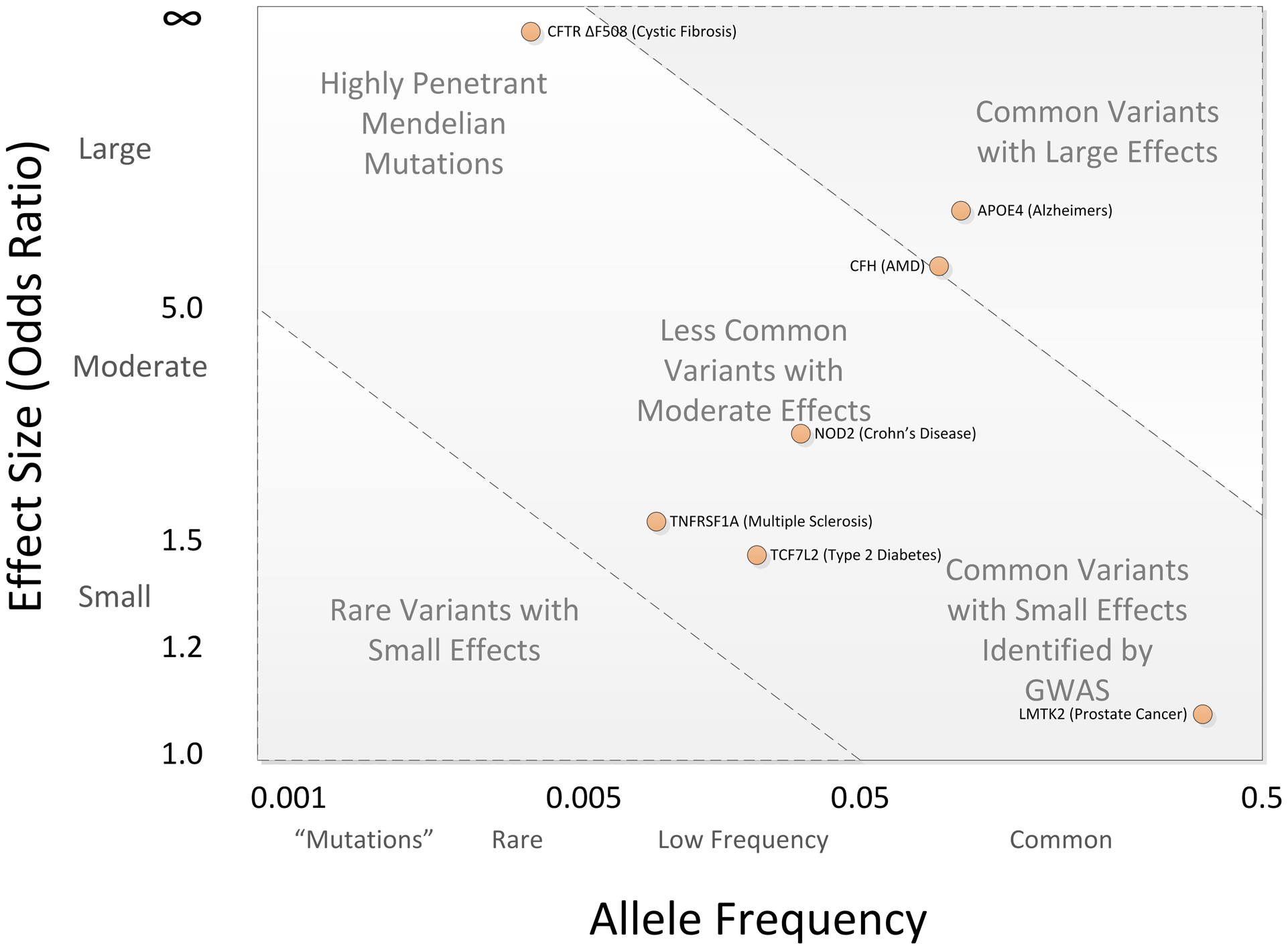
Genome-wide association studies (GWAS) aim to find genetic risk factors for complex phenotypes (Bush et al., 2012). This is achieved by systematically surveying a high number of genetic variants (from hundred thousands to a million). Due to the partial correlations expected, GWAS require large sample sizes, usually above several thousands. Because we are examining considerably more features than samples we have, GWAS is said to be conducted in a low n, high p setting. This, altogether with the small effect sizes, poses a crucial problem: low statistical power (Button, 2013). This has with several implications. First, it implies by definition a small chance of discovering effects that are true. Second, it raises the probability of a discovery to be a false positive. Third, when an underpowered study discovers a true effect, it is likely that the estimated is exaggerated. One practical consequence of this low power in GWAS studies, for example, is the difficulty to reproduce a GWAS result (Visscher, 2008).
In the classical GWAS setting, a statistical test for association is conducted between each variant and the phenotype of interest (Bush et al., 2012). It is usually followed up by a statistical validation by genotyping of the “best hits” in independent samples. Due to its low cost and widespread availability, GWAS has been pushed beyond this realm. Many fields that profit from this ability to survey a sample genome are using GWAS, especially in personalized medicine (e.g. pharmacogenomics). In consequence, GWAS has become a paragon of translational bioinformatics.
The ultimate goal is to be able to identify individuals at genetic risk, as well as understand the molecular basis of the disease. The SNPs associated to the disease can have functional consequences by themselves (e.g. change an amino acid in the protein). However, in the context of genetic studies, SNPs are used as markers of a chromosomic region. In consequence, the SNPs associated with a disease are not necessarily causal: they might just be close and in linkage disequilibrium with the causal variant.
Challenges
Vischer conducted one of the biggest GWAS experiments to date, to inquire about the heritability of height (Visscher, 2008). Three studies with a total sample size of ∼63k individuals (14k + 16k + 34k) identified 54 variants that are reliably associated with height. However, there was an alarmingly low overlap between the causal SNPs identified in the different studies. They proposed two reasons for this. First, we are dealing with SNPs that have a very limited effect size (% of variation explained). In consequence, we need sample sizes of the order of tens of thousands to reliably identify one causal SNP. Second, we are conducting the hundreds of thousands of association tests. Thus, we need to be very stringent with the significant threshold in order to minimize type I errors (false positives).
Another consideration regarding GWAS is that most studies have been carried out on European ancestry populations, which has a reduced variability in comparison to other human populations. In fact, studies on non-European populations have yielded a big number of new, intriguing variants.
References
- Bush, W. S., & Moore, J. H. (2012). Chapter 11: Genome-wide association studies. PLoS Computational Biology, 8(12), e1002822. https://doi.org/10.1371/journal.pcbi.1002822
- Button, K. S., Ioannidis, J. P. A., Mokrysz, C., Nosek, B. A., Flint, J., Robinson, E. S. J., & Munafò, M. R. (2013). Power failure: why small sample size undermines the reliability of neuroscience. Nature Reviews Neuroscience, 14(5), 365–376. https://doi.org/10.1038/nrn3475
- Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., … Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature, 461(7265), 747–753. https://doi.org/10.1038/nature08494
- Visscher, P. M. (2008). Sizing up human height variation. Nature Genetics, 40(5), 489–90. https://doi.org/10.1038/ng0508-489